![]()
![]()
Pharmaceutical companies, wholesalers, parallel importers, and pharmacies are facing European regulatory compliance deadlines that address serialisation of prescription drugs, compliance reporting, and product verification. There is significant work that needs to take place across the entire supply chain. If you aren’t thinking about the full scale and scope of your serialization requirements and business processes to comply to FMD, you may be out of compliance. What follows is an overview of the law, resource requirements, and the serialization solution capabilities you need to meet the FMD requirements.
The EU FMD itself is a relatively brief, 14-page legal document that is directed at the European Union member states. There are currently 28 Member States, including the United Kingdom (UK), which is in the process of leaving the EU, but, interestingly, the UK intends to continue participating in the EU FMD. There are other countries outside the 28 member states that have also announced their intention to participate, including Switzerland, Iceland, Liechtenstein, and Norway. Europe is a big pharmaceutical market, second only to the United States.
The EU FMD was enacted by the EU's European Commission in 2011. It lays out the logical and legal justification for taking Union-wide action against falsified medicines, and then it directs the EU member states to enact standardized regulations to protect the drug supply chain from falsified medicines.
Most importantly, it directs the passage of, a delegated act that requires safety features on prescription drug packages. The FMD did not provide any clue as to what a “safety feature” was, but always referred to them in plural… as in multiple features on each package.
And what are those safety features used for? Here is what the FMD says:
The FMD was enacted in 2011 and it directed the passage of a Delegated Regulation. It took the European Commission four years to develop the Regulation, and it became official on February 9, 2015. The 27-page regulation explains there must be two safety features on each package of prescription drugs and it provides the details of those features. It also explains the System of Repositories in detail, and the obligations and responsibilities of these entities, which are:
And about the costs:
And if that weren't clear enough:
The Delegated Regulation is packed with very useful information, and it is well-written, which makes it fairly, easy to understand.
Figure 1: FMD Timeline
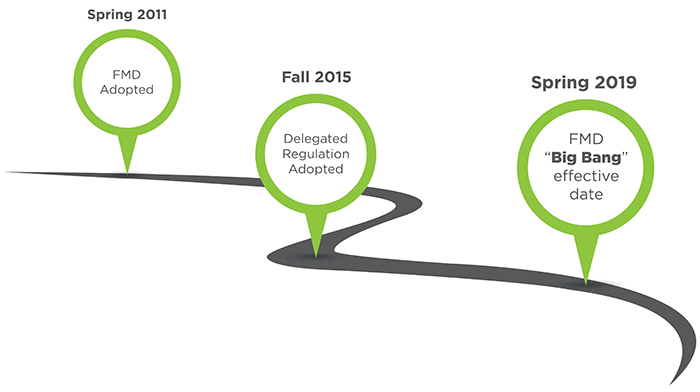
Above is the timeline showing when the original FMD and Delegated Regulation were enacted, and when the “big bang” effective date was. The reason February 9, 2019 is sometimes referred to as a “Big Bang” is to draw a distinction between the way the Drug Supply Chain Security Act (DSCSA) is designed in the United States, and the way the FMD is designed in the EU. The DSCSA is a 10-year-long series of milestones with slowly escalating requirements. In contrast, the FMD and Delegated Regulation really had only one milestone, and that was February 9, 2019. That’s when everything started. All at once. The “big bang”.
As was said above, the Delegated Regulation is where all the details are, and it is well-written and relatively easy to understand. Let’s take a closer look at some of its provisions.
Perhaps one of the most important provisions is the definition of those “safety features”. There are just two. One is, an anti-tamper device. The Delegated Regulation defines it as:
And surprisingly, that’s all it says about it. You are left to figure it out, but it is probably safe to say these are the kind of features that have been used on Over-The-Counter drug packaging ever since the Tylenol poisonings that occurred in the US in the mid-1980’s.
Examples include heat-shrink, seals, glue, and tape. Anything that can’t be put back after opening the package.
The second safety feature is much more interesting. It’s the Unique Identifier. The Delegated Regulation defines it as
In contrast with the anti-tamper device, there is a lot written in the Delegated Regulation about this safety feature, directly and indirectly. The Unique Identifier must be composed of a product code, serial number, national reimbursement number (where required), batch number, and expiration date. It must be carried within a 2D Data Matrix barcode and in human-readable text on the package.
The serial number data element is defined as up to 20 alphanumeric characters “...generated by a deterministic or a nondeterministic randomization algorithm…,” with the odds of guessing a valid one, being less than 1 in 10,000. This randomization requirement was originally unique to the FMD, although it is being emulated elsewhere. In the DSCSA, there is no such requirement. And that makes more sense in a point-of-dispense authentication system like the FMD than it does in a regulation like the DSCSA. But randomization is also one of the complications in this Regulation.
Another important concept described in detail in the Delegate Regulation is the System of Repositories.
Figure 2. System view of the FMD “System of Repositories”, source: EMVO
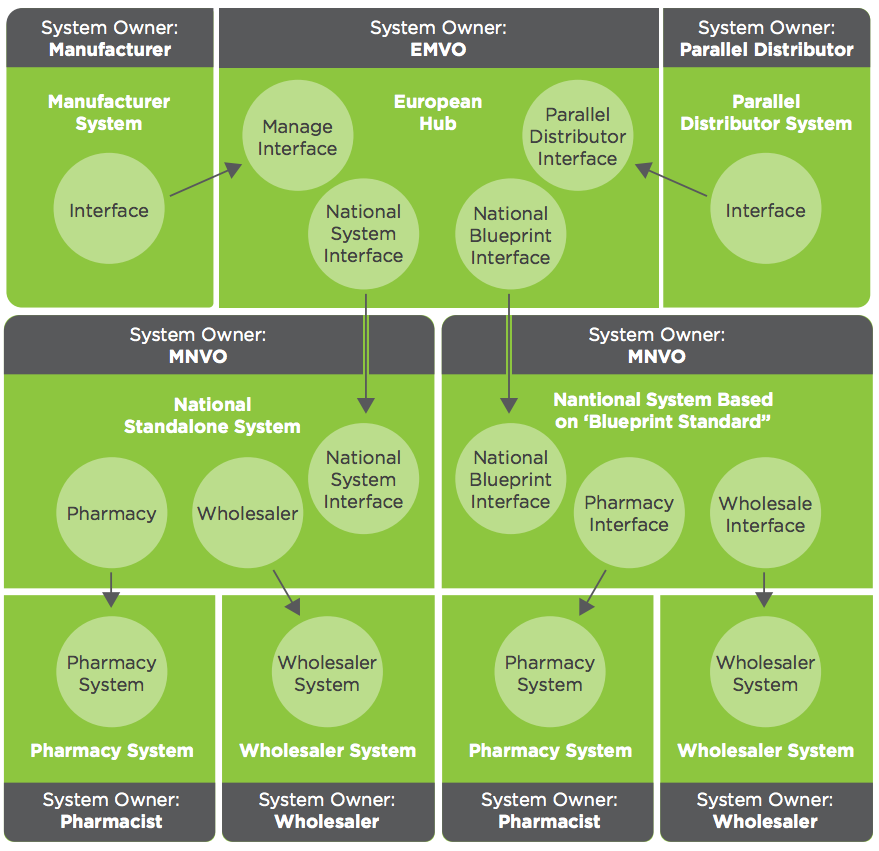
Above is a logical view of the System of Repositories, provided by the European Medicines Verification Organization (EMVO). The top center box is what is known as the European Hub, which has been built by the EMVO, and is the primary place drug manufacturers and parallel distributors will need to send data about the products they introduce into the supply chain. You can see those two entities represented in the upper left and upper right boxes.
The EU Hub retains all the master data about each product, and it passes all of the product data it receives—including the master data and serial numbers—to the national system repositories that are associated with the regions where the drugs can be marketed. Usually, this is a subset of the European Union. Each region maintains their own national repository, whether it is a standalone system, or one based on the EMVO national blueprint. Most of them follow the EMVO blueprints.
A national repository is usually operated by a single member state, but there are some that are shared between multiple states. As you can see in the lower row of boxes in Figure 2, wholesale distributors and pharmacies need to connect to each of the national repositories that serve the regions where they are located. When they need to verify a given product, they must submit a request to one national repository that serves the region where the drug is being dispensed or distributed.
That’s how the System of Repositories is built and used, but there are a few additional details. One is, if a given drug manufacturer or parallel distributor only markets drugs in a single region of the EU, they can send their data directly to that specific national repository, bypassing the EU Hub. Another is, if the national repository cannot find a record matching the Unique Identifier being verified by a wholesale distributor or pharmacy, the national repository can send a request to the EU Hub to find out which national repositories hold the verification data for it.
Under the FMD, drug manufacturers have certain obligations. First, they must begin applying the safety features to each drug package. As we’ve seen, this includes the antitamper device and the Unique Identifier.
And they must send the data to the EU Hub. That data includes:
Verification of the safety features is a fundamental process under the FMD. Here are some extracts from the Delegated Regulation which describe what is meant by “Verification”.
This is important for patient safety. In the EU, “verification” means you must check both things. To be able to check the integrity of the anti-tamper device, you must be able to see the individual unit. This means you can’t verify an entire case or pallet just by knowing the Unique Identifiers on the individual packages they contain. However, these two verification steps can be performed at different times, and the check of the integrity of the anti-tamper device is most important right before the drug is provided to a patient.
Wholesalers and repackagers have their own set of obligations under the FMD. They are obligated to verify drugs that have the safety features on them, under these conditions:
Decommissioning is another very important concept under the FMD. It’s a way of indicating in the System of Repositories that a given Unique Identifier is no longer valid, or at least, should not be dispensed to a patient. The Delegated Regulation says that decommissioning should set the drug to an inactive status in the system of repositories.
Wholesale distributors and repackagers are required to decommission in these situations:
And, whenever products are being distributed to certain smaller entities, like schools, individual practitioners, veterinarians, optometrists, police, military, prisons, etc. These are entities that are not expected to perform the verification for themselves, so to ensure that a verification is done before dispensing or administration to a patient, the law expects the wholesaler or repackager to do it.
Finally, persons authorized or entitled to supply medicinal products to the public, or dispensers, also have specific obligations under the FMD.
They are required to verify and decommission drugs under these situations:
Now let’s look at some of the complications for drug manufacturers when meeting the FMD, and particularly the Delegated Regulation.
First, the randomization of the serial number is a significant difference from most regulations prior to the FMD. Make sure your serialization solution system can meet the 1 in 10,000 guessing requirements.
The next complication is the “Big Bang” startup of the regulation. Overnight, on February 9, 2019, everyone in the EU pharma supply chain had to start fulfilling the requirements spelled out for them in the Delegated Regulation. The manufacturers began applying Unique Identifiers to their product packaging and upload the data to the EU Hub. And, downstream trading partners began verifying and, in some cases, decommissioning products that have an FMD Unique Identifier on them.
Verification is another complication. In some cases, it must be done without decommissioning, and in other cases, it must be done along with decommissioning. Here are the primary situations where each is necessary.
Verification
Decommissioning
If a downstream trading partner mistakenly decommissions a set of unique identifiers, they can “re-commission” them. There are restrictions, but this could get complicated for some. Decommissioning and re-commissioning will mostly be done by downstream trading partners. If they fail to do it properly, the manufacturer may end up having to deal with collections of good product that cannot be dispensed because it is mistakenly indicated as decommissioned in the System of Repositories.
Whenever decommissioning is necessary on a full case, full pallet, or full shipment, as it is during an export, or when a full trailer is hijacked or destroyed, companies would benefit from having aggregation data. That way they do not have to open every case and scan each unit, which is something you cannot do anyway if the drugs are stolen. Unfortunately, the FMD and Delegated Regulation do not anticipate this need, so aggregation data is not required. Not only that, but the EU Hub and the System of Repositories do not even recognize aggregation data, so if it is created voluntarily, companies have to share it on their own.
The FMD and its point-of-dispense authentication model was originally touted as being much easier than full track and trace through the supply chain because it did not need aggregation data, among other things. But this has become an issue now that companies are starting to run into these problems.
Another complication is the handling of free samples. In the EU, free samples must be serialized right along with every other product. When the product is later converted to free samples, either by the manufacturer, the wholesale distributor, or the dispenser, the Unique Identifiers must be decommissioned in the System of Repositories. That way, if they end up back in the supply chain they won’t verify, so they should not be dispensed or administered. In contrast, most other regulations, including the DSCSA, do not even require free samples to be serialized.
The FMD is profoundly changing the pharma industry in Europe. Companies need to align themselves with vendors who have a depth of experience with serialization and meeting pharma traceability regulations all around the world.
©2025 Systech. All rights reserved.